

")
")
")
")

GPCRs are hot targets in drug discovery, with approximately 30% of marketed drugs acting upon them. As a representative member of the GPCR family, AT1R activates the Gq protein signaling pathway upon binding its endogenous ligand, Angiotensin II (AngII), triggering physiological responses like vasoconstriction. Dysregulation of AT1R is directly linked to various cardiovascular diseases, including hypertension and cardiac hypertrophy. Although Angiotensin Receptor Blockers (ARBs) are widely used as antihypertensive drugs, traditional antagonists suffer from mechanisms that are often singular and exhibit variable patient response.
More critically, GPCR activation exhibits "biased signaling," where different ligands can selectively activate specific signaling pathways. For instance, studies have shown that β-arrestin-biased AT1R agonists can improve cardiac contractility without affecting blood pressure, offering a new direction for heart failure treatment. However, the conformations of peptide-activated GPCRs are highly dynamic and difficult to crystallize. The lack of high-resolution structures of their active states has prevented a clear understanding of ligand-receptor interaction details and the mechanism of action of biased agonists. Previously reported AT1R crystal structures were all in the inactive state bound to antagonists, which have fundamentally different binding modes compared to peptide agonists. There was a pressing need to obtain the active-state structure to fill this critical knowledge gap.
This study marks the first successful determination of the crystal structure of the human AT1R in its active state, at a resolution of 2.9 Å. This structure reveals the detailed binding mode of AT1R with a partial agonist peptide, S1I8, and showcases a receptor conformation stabilized by a synthetic nanobody (Nb.AT110i1). The research not only elucidates the unique activation mechanism of AT1R but also highlights its significant differences from the activation modes of other known GPCRs, providing a structural basis for understanding biased signal transduction.
合成纳米抗体的筛选与优化Screening and Optimization of Synthetic Nanobodies
The dynamic and unstable nature of GPCR conformations is a major obstacle to crystallization studies. Nanobodies, with their small size and long antigen-binding loops, serve as ideal tools for stabilizing target conformations. However, traditional immunization methods failed to yield AT1R-specific nanobodies. The team therefore turned to a synthetic yeast surface display nanobody library for screening.
The screening process involved two main steps: first, two rounds of Magnetic-Activated Cell Sorting (MACS) were used to enrich for AT1R-binding nanobodies. This was followed by Fluorescence-Activated Cell Sorting (FACS) to isolate conformation-specific clones. Yeast cells were co-incubated with AT1R bound to the inverse agonist olmesartan (labeled with AF488) and AT1R bound to the agonist AngII (labeled with AF647), allowing selective collection of yeast clones binding specifically to the active-state AT1R. This process yielded the highly specific nanobody Nb.AT110. Research confirmed that it co-immunoprecipitates with AT1R only in the presence of an agonist and can enhance the affinity of AngII for AT1R by approximately 6-fold.
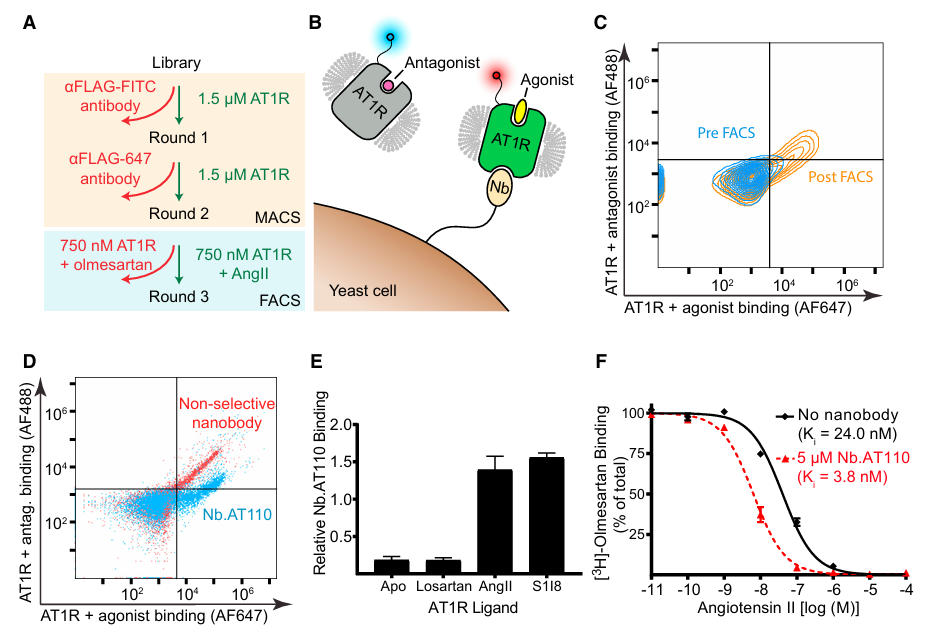
Figure 1: Screening for Active-State AT1R Specific Nanobodies using Yeast Surface Display
To meet the requirements for crystallization, the team constructed a mutant library via error-prone PCR. After two rounds of FACS-based affinity maturation, they selected the variant Nb.AT110i1, which contains four key mutations: A31V, N58D, I98V, and Y113N (Figure 2). This variant exhibited an over 100-fold improvement in affinity for the active-state AT1R compared to the original nanobody, laying the foundation for subsequent complex crystallization.
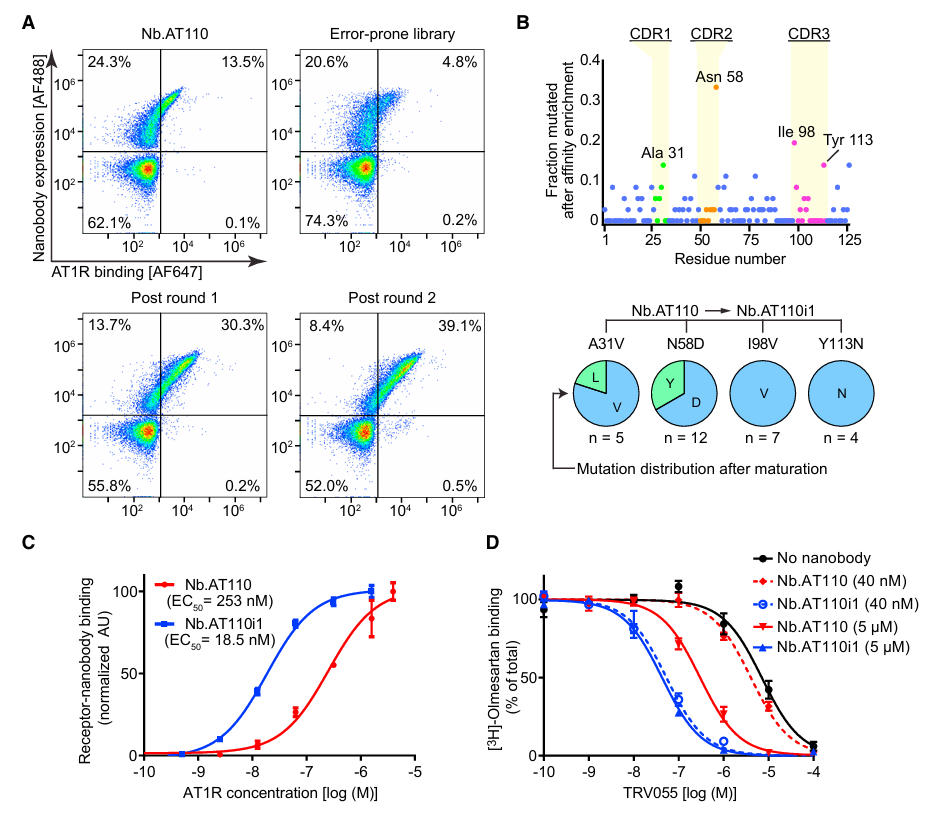
Figure 2: Affinity Maturation of Nb.AT110
Crystallization and Structure Determination of the AT1R-Ligand-Nanobody Complex
To address the issue of low ligand affinity in traditional AT1R crystallization constructs, the team redesigned the protein expression and crystallization construct. They inserted the thermostabilized apocytochrome b562 RIL (BRIL) into the third intracellular loop (ICL3), preserved the intact N-terminal sequence to avoid affecting peptide ligand binding, and performed C-terminal truncation. This construct demonstrated a 2-fold increase in affinity for AngII compared to the wild-type receptor and could stably bind Nb.AT110i1.
The study used the AngII analog S1I8 as the ligand, forming a ternary complex with AT1R and Nb.AT110i1. Diffraction-quality crystals were obtained using the lipidic cubic phase crystallization method. By merging diffraction data from 5 crystals, the structure was solved by molecular replacement at 2.9 Å resolution. The structure revealed that Nb.AT110i1 binds to the intracellular transducer pocket of AT1R, with its exceptionally long CDR3 loop inserting into the receptor core, stabilizing the characteristic conformational changes of the active state.
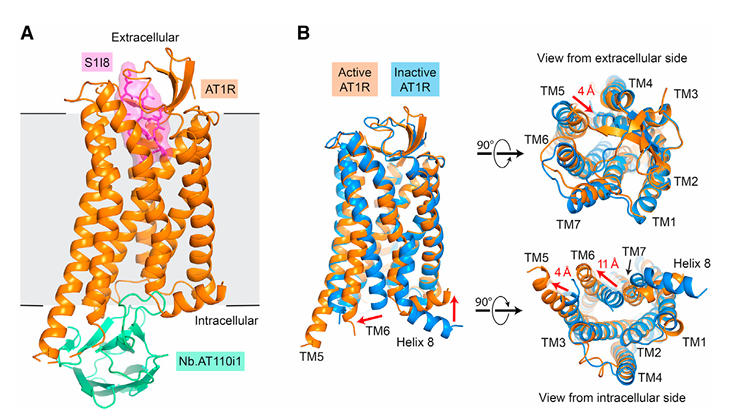
Figure 3: Active-State AT1R Structure Stabilized by Nb.AT110i1
Revealing the AT1R Activation Mechanism and Ligand Binding Mode
Structural analysis revealed the uniqueness of the AT1R activation mechanism: S1I8 binds to the receptor pocket in an extended conformation, with its N-terminus facing the extracellular solvent and its C-terminus buried deep within the receptor core, forming an extensive interface through polar and hydrophobic interactions.
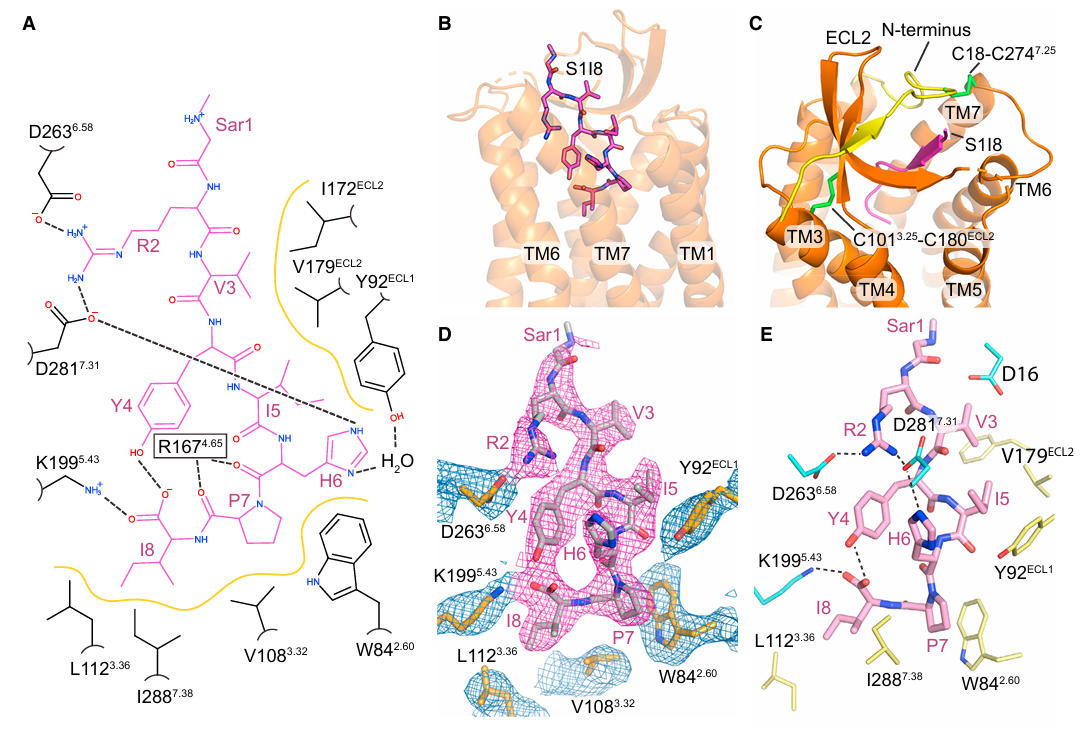
Figure 4: Binding Mode of S1I8 to AT1R
The interaction between the C-terminal I8 residue of the ligand and receptor residues like K1995.42 initiates a conformational cascade: W2536.48 and Y2927.43 shift downwards, triggering a rearrangement of the phenylalanine ratchet between TM5 and TM6. This ultimately leads to an outward displacement of TM6 by 11 Å, forming the G protein binding site.
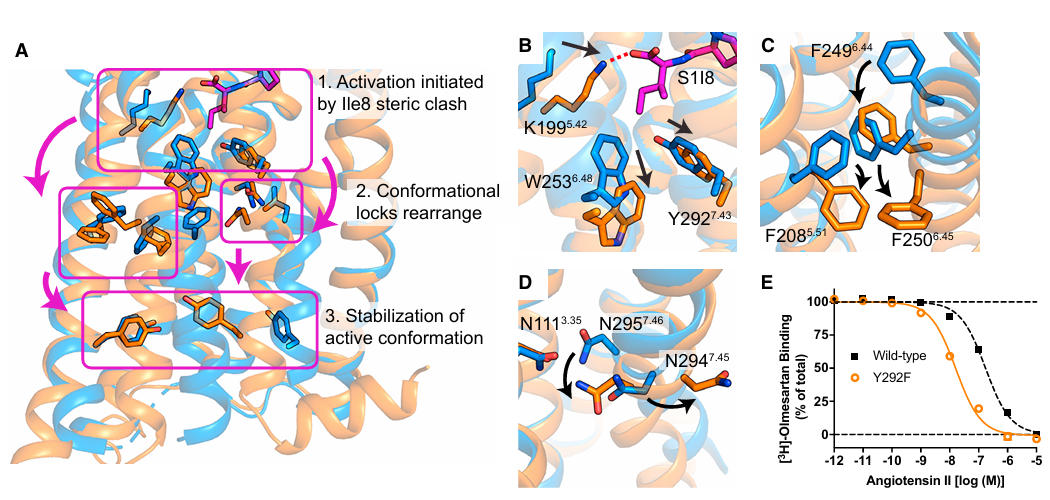
Figure 5: Activation Mechanism of AT1R
Unlike other GPCRs, AT1R stabilizes its inactive state via a hydrogen bond between N1113.35 and N2957.46, effectively replacing the role of a sodium ion. Furthermore, the active-state binding pocket is significantly constricted due to inward movements of TM5 and TM7, creating a clear distinction from the antagonist-bound inactive state pocket. This explains the inverse agonism property of ARBs – meaning they not only block agonist action but also suppress the receptor's basal activity. Additionally, S1I8 forms a unique partial β-barrel structure with the AT1R's N-terminus and ECL2, where the peptide chain and receptor fragments together form an incomplete barrel-like structure, a binding mode relatively unique among peptide-binding GPCRs. This finding provides new insights into the ligand recognition mechanisms of peptidergic GPCRs.
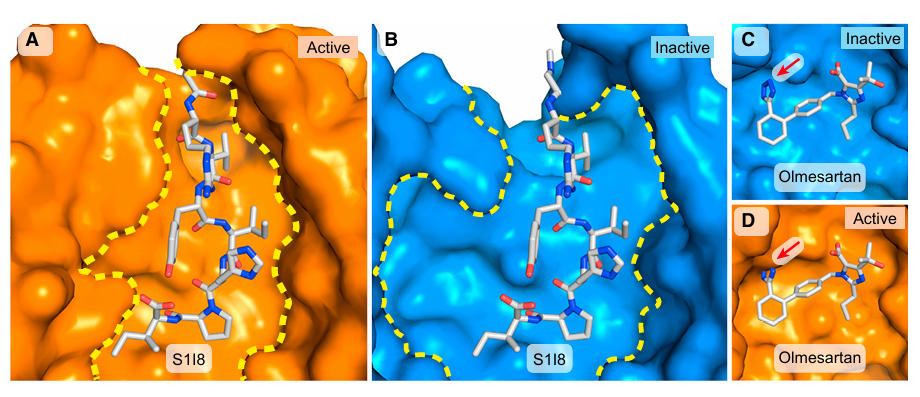
Figure 6: Differences Between the Active and Inactive State AT1R Binding Pockets
Validation of the Structural Basis for Biased Signaling
DEER spectroscopy confirmed that the conformational ensemble of AT1R is closely related to the ligand type: the conformation stabilized by the full agonist AngII closely matches the crystal structure, whereas β-arrestin-biased agonists stabilize incompletely activated conformations. The structure shows that the volume of the ligand's C-terminal residue is key to the bias. The phenyl ring of F8 induces a more extensive conformational rearrangement in the receptor core, whereas smaller side chains cannot fully trigger this process, leading to signaling bias towards the β-arrestin pathway. This discovery provides a clear structural target for designing highly selective biased drugs.
This study not only reveals the active structure of AT1R for the first time but also demonstrates the powerful potential of synthetic nanobodies in GPCR structural biology. Nb.AT110 and its variants bind AT1R only in the presence of an agonist, showing no cross-reactivity with the inactive receptor or other GPCRs. This precise targeting ensures the stable capture of the active conformation, solving the core problem of conformational heterogeneity in GPCR structural research. As such tools and methods become more widespread, we can expect to see the structures of more "elusive" membrane proteins solved, bringing revolutionary breakthroughs to drug discovery and disease treatment.

Wuhan Nano Body Life Science and Technology Co. Ltd. (NBLST) is a nanobody industry platform established under the initiative of the Wuhan Industrial Innovation and Development Research Institute. Its headquarters is located in the main building of the Wuhan Industrial Innovation and Development Research Institute in the East Lake High-tech Development Zone, Wuhan. It boasts a 1400 m² independent laboratory in the Precision Medicine Industrial Base of Wuhan Biolake. Additionally, NBLST has established alpaca experimental and transfer bases in Zuoling, Wuhan, and Tuanfeng, Huanggang, both compliant with laboratory animal standards. These bases currently house over 600 alpacas, providing "zero-immunization-background" guaranteed alpaca immunization services for research institutions and antibody drug development companies.
NBLST focuses on the development, engineering, and application of nanobodies, and is dedicated to building an integrated public experimental service platform for production, education, and research. It possesses a full-chain technology platform encompassing antigen preparation (peptides, proteins, and RNA), antibody discovery and engineering, through to biological function validation/screening. The RNA antigens include RNA structurally and sequentially optimized for alpacas. Antibody discovery and engineering services employ multiple technological routes, including phage display, RNA, and mammalian cell display. Through cross-complementation of multiple platforms, it provides flexible antibody discovery and engineering services for pharmaceutical companies and research institutes, facilitating the development of drug reagents.
In addition to its natural nanobody library, NBLST also offers an off-the-shelf immunized library to help clients quickly screen for antibody molecules that meet their needs.
If you require our services, please feel free to contact us via email: marketingdept@nanobodylife.com